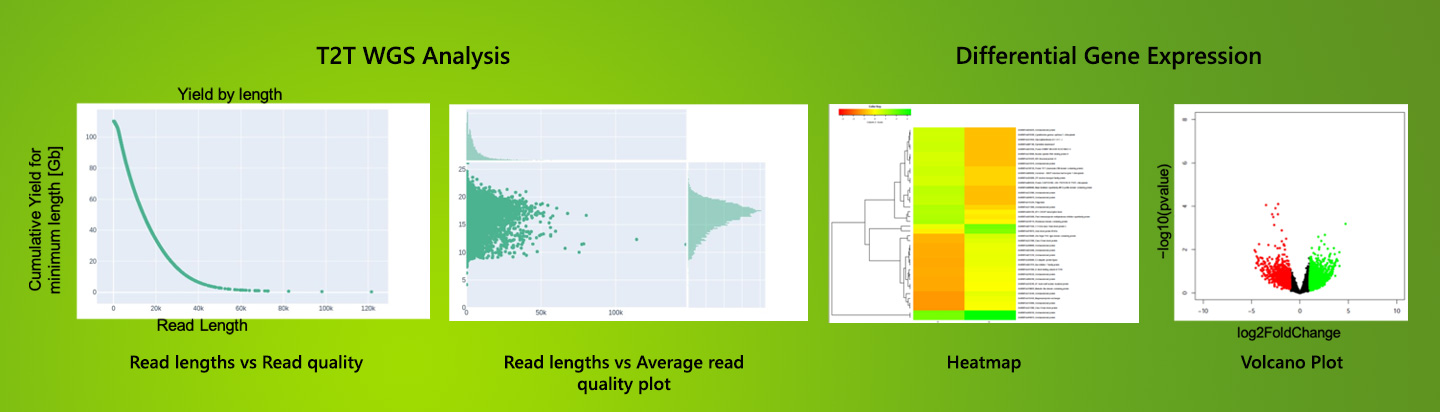

Whole Genome Sequencing
(WGS)
WGS involves the sequencing of entire genome of a organism both protein and
non-protein coding (including regulatory) region of the genome. Using long
read
technology we can accomplish telomere-to-telomere (T2T) de novo assembly of
the
genome without gaps and errors. At QTLomics long read high-output PromethION
Nanopore Sequencing platform is used for WGS projects.

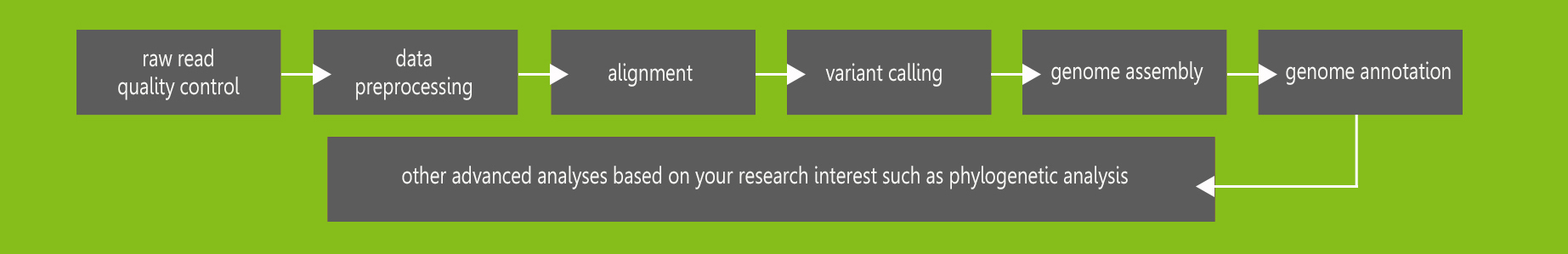
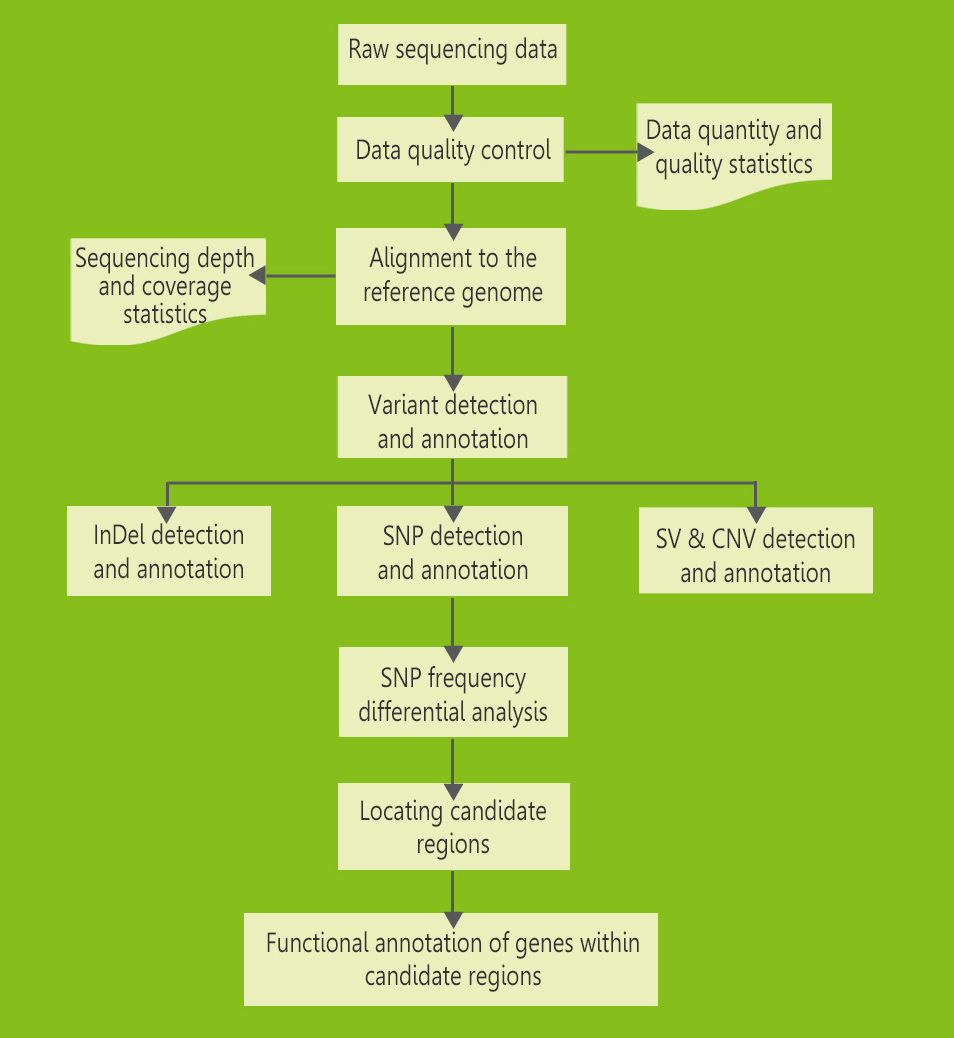
Bioinformatics workflow
The bioinformatics workflow for WGS is similar to that for whole exome
sequencing. You can view our article Bioinformatics Workflow for Whole Exome
Sequencing.
The bioinformatics workflow for WGS falls into the following steps:
Transcriptomics sequencing
Transcriptome sequence analyses provides information about differentially
expressed genes between samples, alternative splicing events, the detection
of
single nucleotide polymorphisms (SNPs) and insertions/deletions (InDels),
making
it an essential tool for various biological studies. We apply both Nanopore
and
Illumina platforms for transcriptome sequencing.
Transcriptome Sequencing
Oxford Nanopore Bioinformatics Steps
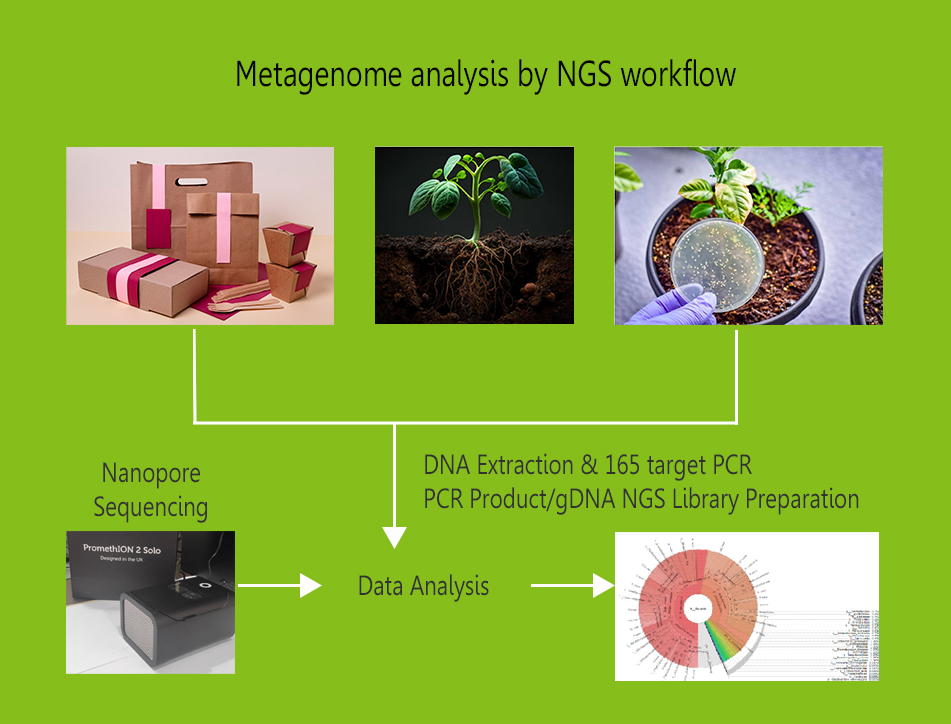
Metagenomics would analyse the totality of the genomic material
present
in a sample.
Whole genome metagenomics:
WGS
metagenomics aims to sequence all genomes
existing in an environmental sample to analyse the biodiversity and
the
functional capabilities of the microbial community studied.
In WGS metagenomics the entire genetic material of a sample is
recovered, which enables the characterization of the complete
diversity
of a habitat, including archaea, bacteria, eukaryotes, viruses, and
plasmids, as well as its gene content.
16S/ITS targeted
sequencing: 16S rRNA
sequencing is applied to identify
bacteria down to the genus and species level. The internal
transcribed
spacer 1 (ITS1) region of the rRNA cistron is used as a DNA marker
for
identification of fungal species in complex samples.

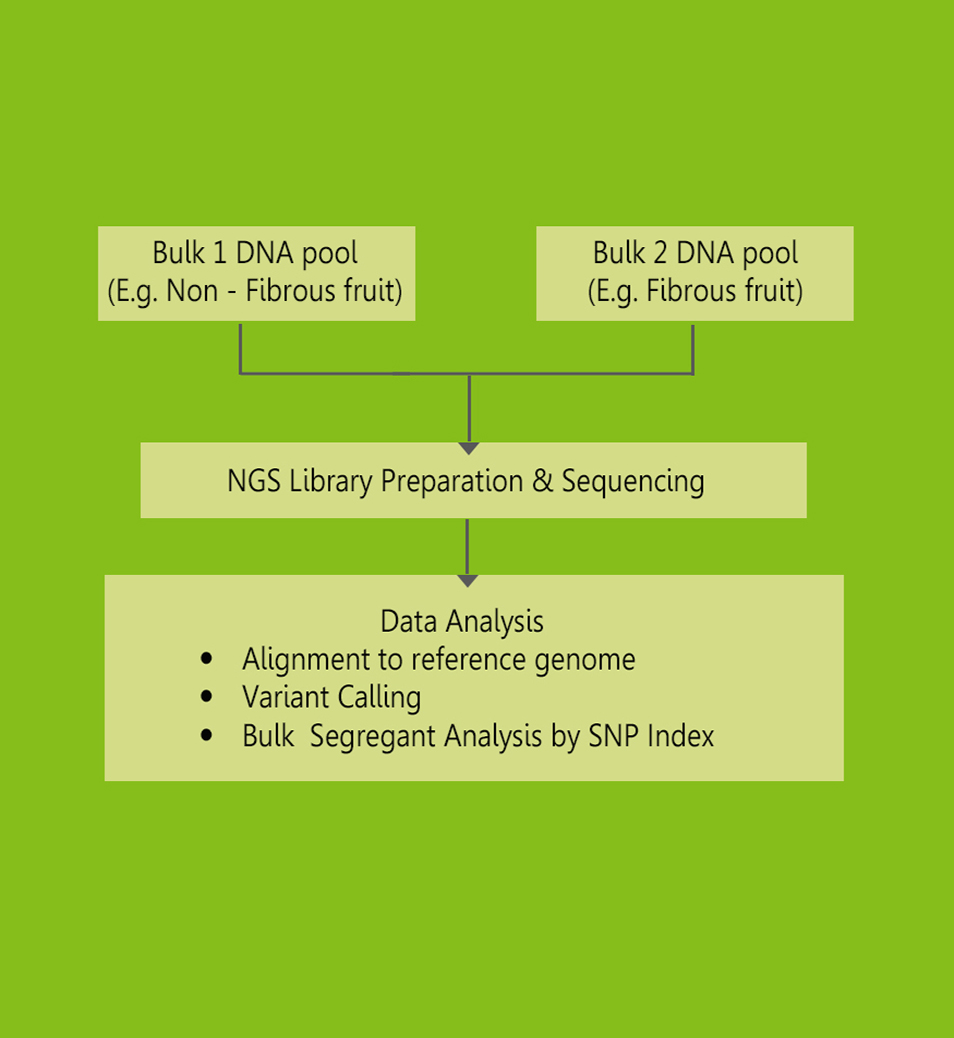
Bulked-segregant analysis (BSA) by NGS is an elegant method to identify DNA markers tightly linked to the causal gene for a given phenotype. Majorly for the agronomically crop and controlled by the QTL.Mapping and isolation of QTLs is important for efficient crop breeding by marker-assisted selection (MAS) and for a better understanding of the molecular mechanisms underlying the traits.
 Workflow
Workflow
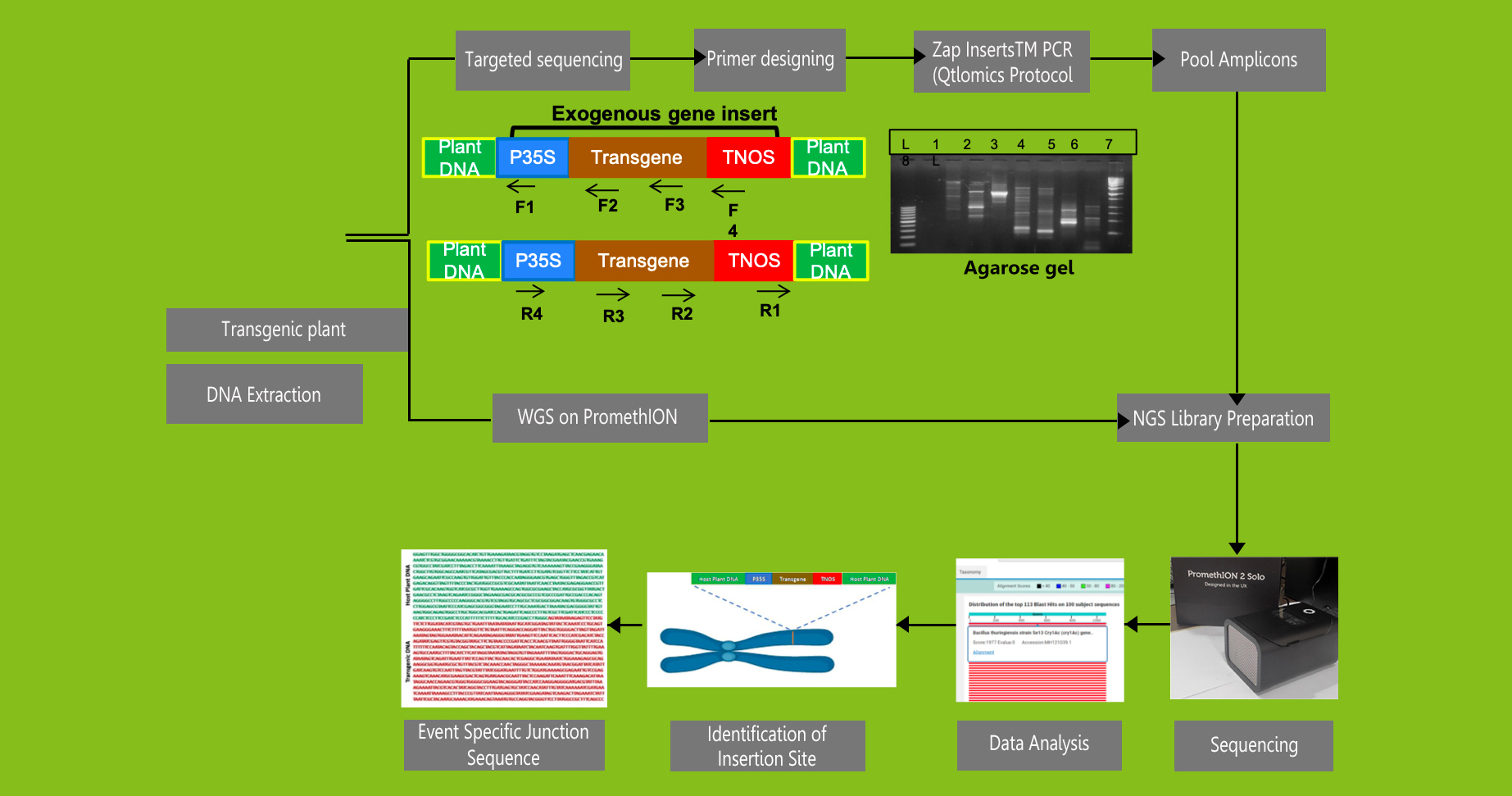
Herbal industry suffers from substitution and adulteration of medicinal
herbs with closely related species. The
efficacy of the drug decreases if it is adulterated, and in some cases, can
be lethal if it is substituted with
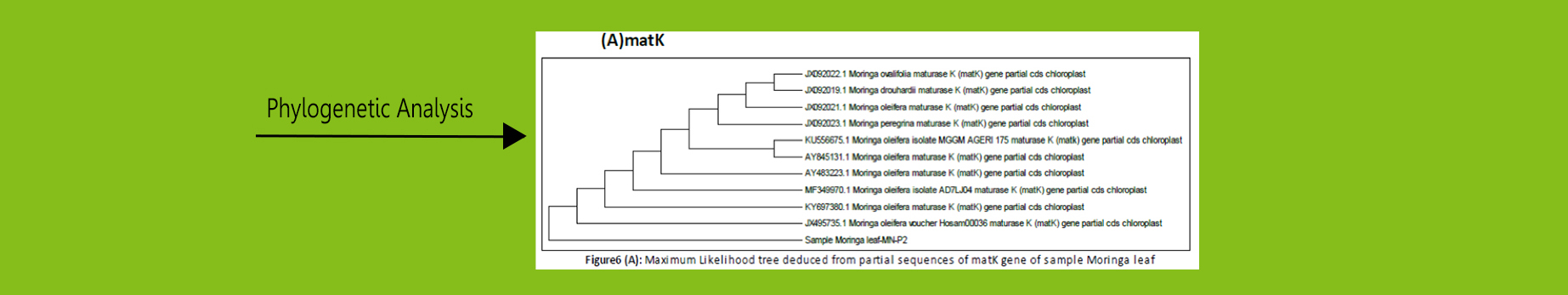
toxic adulterants. DNA barcoding is the valuable tool for the Identification
of medicinal plants
DNA barcoding uses specific regions of DNA in order to identify species.
Well known genomic regions (matK, ITS,
psbA-trnH, rbcL) were used for DNA Barcoding in plants.

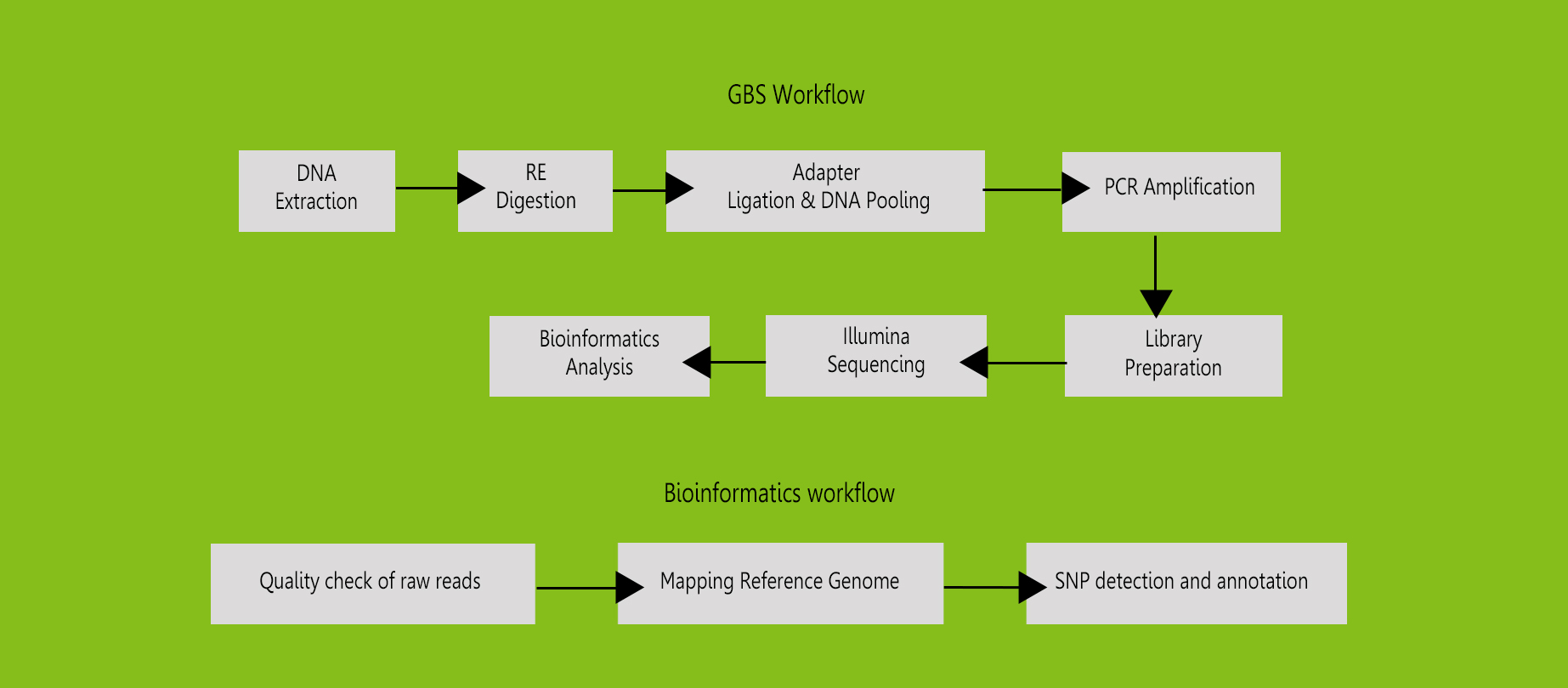
GBS is a novel application of NGS protocols for high throughput discovery of
SNPs and simultaneous
genotyping in multiple DNA samples. It is also applied for genome-wide
association study (GWAS), genomic
diversity study, genetic linkage analysis, molecular marker discovery and
genomic selection under a large
scale of plant breeding programs. GBS is becoming increasingly important as
a cost-effective and unique tool
for genomics-assisted breeding in a range of plant species.